DTINet is a computational pipeline to predict novel drug-target interactions (DTIs) from heterogeneous network. DTINet focuses on learning a low-dimensional vector representation of features for each node in the heterogeneous network, and then predicts the likelihood of a new DTI based on these representations via a vector space projection scheme.
You can download the source code from here
Related Papers:
Yunan Luo#, Xinbin Zhao#, Jingtian Zhou#, Jinling Yang, Yanqing Zhang, Wenhua Kuang, Jian Peng*, Ligong Chen* and Jianyang Zeng*.
A Network Integration Approach for Drug-Target Interaction Prediction and Computational Drug Repositioning from Heterogeneous Information.
Nature Communications. 2017.
We propose a deep learning based framework, called ROSE (RibosOme Stalling Estimator), to address the aforementioned challenges and model translation elongation dynamics based on high-throughput ribosome profiling data.
Sai Zhang#, Hailin Hu#, Jingtian Zhou#, Xuan He, Tao Jiang, Jianyang Zeng*. Analysis of ribosome stalling and translation elongation dynamics by deep learning. Cell Systems. 2017. In press.
TITER is a deep learning-based framework, for accurately predicting TISs on a genome-wide scale based on QTI-seq data. TITER extracts the sequence features of translation initiation from the surrounding sequence contexts of TISs using a hybrid neural network and further integrates the prior preference of TIS codon composition into a unified prediction framework.
Particle picking is a time-consuming step in single-particle analysis and often requires significant interventions from users, which has become a bottleneck for future automated electron cryo-microscopy (cryo-EM). Here we developed a deep learning framework, called DeepPicker, to address this problem and fill the current gaps toward a fully automated cryo-EM pipeline. DeepPicker employs a novel cross-molecule training strategy to capture common features of particles from previously-analyzed micrographs, and thus does not require any human intervention during particle picking.
Characterizing the binding behaviors of RNA-binding proteins (RBPs) is important for understanding their functional roles in gene expression regulation. However, current high-throughput experimental methods for identifying RBP targets, such as CLIP-seq and RNAcompete, usually suffer from the false negative issue. Here, we develop a deep boosting based machine learning approach, called DeBooster, to accurately model the binding sequence preferences and identify the corresponding binding targets of RBPs from CLIP-seq data.
Shuya Li#, Fanghong Dong#, Yuexin Wu#, Sai Zhang, Chen Zhang, Xiao Liu, Tao Jiang, Jianyang Zeng*.
A deep boosting based approach for capturing the sequence binding preferences of RNA-binding proteins from high-throughput CLIP-seq data.
Nucleic Acids Research. 2017.
GEM (Genomic organization reconstructor based on conformational Energy and Manifold learning) is a novel manifold learning framework, to elucidate the underlying 3D
spatial organizations of chromosomes from Hi-C data. Unlike previous chromatin structure
reconstruction methods, which explicitly assume specific relationships between Hi-C
interaction frequencies and spatial distances between distal genomic loci, GEM is able
to reconstruct an ensemble of chromatin conformations by directly embedding the neighboring
affinities from Hi-C space into 3D Euclidean space based on a manifold learning
strategy that considers both the fitness of Hi-C data and the biophysical feasibility of the
modeled structures, which are measured by the conformational energy derived from our
current biophysical knowledge about the 3D polymer model.
gOSPREY is the abbreviation for Open Source
Protein REdesign for You on a
GPU. It enables the user to utilize a GPU to accelerate
the procedure of protein design in a massive parallel fashion.
gOSPREY is based on the software suite OSPREY, developed by the
Donald Lab
from Duke University. gOSPREY is a free software and
distributed under GNU General Public License v2.0. You can read the
document and download the source code from
here or from the
Donald Lab.
Related Papers:
Yichao Zhou, Wei Xu, Bruce R. Donald, and Jianyang Zeng*. An Efficient Parallel Algorithm for Accelerating Computational Protein Design. Proceedings of 21st Annual International Conference on Intelligent Systems for Molecular Biology (ISMB), Bioinformatics, 2014.
RDC-PANDA is a suite of programs for nuclear Overhauser effect (NOE) assignment and high-resolution structure determination starting with a global fold calculated from exact solutions to the residual dipolar coupling (RDC) equations. RDC-PANDA is free software and distributed open-source under the GNU Lesser General Public License (Gnu, 2002). The binary executable files, source code and documents of RDC-PANDA can be downloaded here.
Related Papers:
Jianyang Zeng, Kyle E. Roberts, Pei Zhou, Bruce R. Donald. A Bayesian Approach for Determining Protein Side-Chain Rotamer Conformations Using Unassigned NOE Data. Journal of Computational Biology. 2011.
Chittaranjan Tripathy, Jianyang Zeng, Pei Zhou, Bruce R. Donald. Protein Loop Closure using Orientational Restraints from NMR Data. Proteins: Structure, Function, and Bioinformatics. 2011.
Jianyang Zeng, Kyle E. Roberts, Pei Zhou, Bruce R. Donald. A Bayesian Approach for Determining Protein Side-Chain Rotamer Conformations Using Unassigned NOE Data. International Conference on Research in Computational Molecular Biology (RECOMB'11). Vancouver, 2011.
Chittaranjan Tripathy, Jianyang Zeng, Pei Zhou, Bruce R. Donald. Protein Loop Closure using Orientational Restraints from NMR Data. International Conference on Research in Computational Molecular Biology (RECOMB'11). Vancouver, 2011.
Jianyang Zeng, Jeffrey Boyles, Chittaranjan Tripathy, Lincong Wang, Anthony Yan, Pei Zhou, Bruce R. Donald. High-Resolution Protein Structure Determination Starting with a Global Fold Calculated from Exact Solutions to the RDC Equations. Journal of Biomolecular NMR. 45(3):265-281. 2009.
Bruce R. Donald and Jeffrey Martin. Automated NMR Assignment and Protein Structure Determination using Sparse Dipolar Coupling Constraints. Progress in Nuclear Magnetic Resonance Spectroscopy, 55(2):101-127, 2009.
Jianyang Zeng, Chittaranjan Tripathy, Pei Zhou, Bruce R. Donald. A Hausdorff-Based NOE Assignment Algorithm Using Protein Backbone Determined from Residual Dipolar Couplings and Rotamer Patterns. Proceedings of the 7th Annual International Conference on Computational Systems Bioinformatics (CSB'08), Stanford CA, pp. 169-181, August 26-29, 2008.
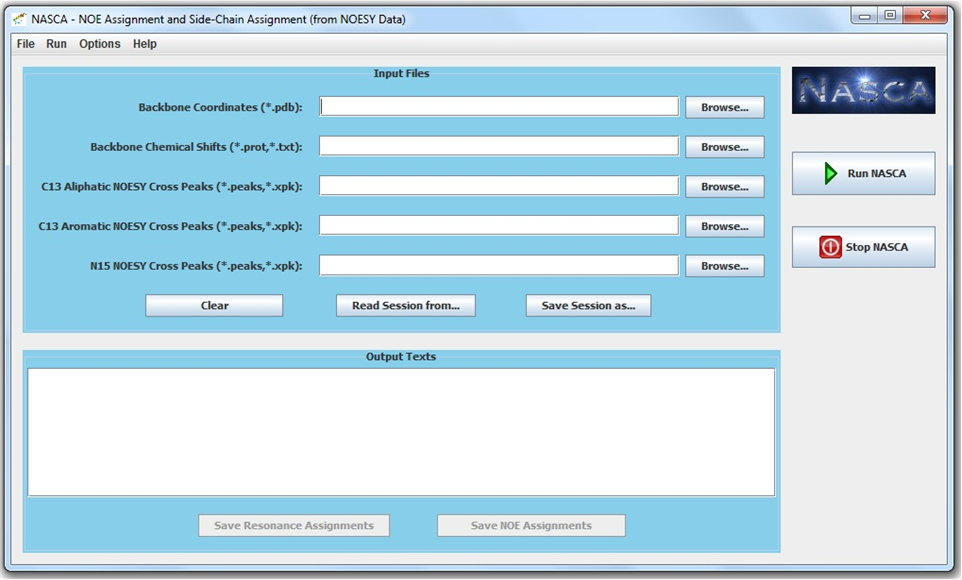
NASCA is an automated program for side-chain resonance assignment and nuclear Overhauser effect (NOE) assignment from NOESY data. It does not require data from TOCSY experiments. NASCA is integrated with a graphical user interface (GUI). NASCA is free software and distributed open-source under the GNU Lesser General Public License (Gnu, 2002). The binary executable files, source code and documents of NASCA can be downloaded here.
Related Papers:
Jianyang Zeng, Pei Zhou, Bruce R. Donald. Protein Side-Chain Resonance Assignment and NOE Assignment Using RDC-Defined Backbones without TOCSY Data. Journal of Biomolecular NMR. 2011.
Jianyang Zeng, Pei Zhou, Bruce R. Donald. A Markov Random Field Framework for Protein Side-Chain Resonance Assignment. International Conference on Research in Computational Molecular Biology (RECOMB'10). Lisbon, Portugal. April, 2010.